Experimental and computational evaluation of the barrier to torsional rotation in a butadiyne-linked porphyrin dimer
Abstract
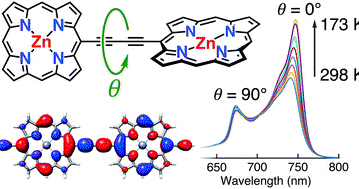
The barrier to torsional rotation in a butadiyne-linked porphyrin dimer has been determined in solution using variable temperature UV-vis-NIR spectroscopy: ΔH = 5.27 ± 0.03 kJ mol−1, ΔS = 10.69 ± 0.14 J K−1 mol−1. The value of ΔH agrees well with theoretical predictions. Quantum chemical calculations (DFT) were used to predict the torsion angle dependence of the absorption spectrum, and to calculate the vibronic fine structure of the S0 → S1 absorption for the planar dimer, showing that the absorption band of the planar conformer has a vibronic component overlapping with the 〈0textbar0〉 absorption of the perpendicular conformer. The torsion barrier in the porphyrin dimer is higher than that of 1,4-diphenylbutadiyne (calculated ΔH = 1.1 kJ mol−1). Crystallographic bond lengths and IR vibrational frequencies confirm that there is a greater contribution of the cumulenic resonance form in butadiyne-linked porphyrin dimers than in 1,4-diphenylbutadiyne. The DFT frontier orbitals of the twisted conformer of the porphyrin dimer are helical, when calculated in the absence of symmetry. The helical character of these orbitals disappears when D2d symmetry is enforced in the 90° twisted conformer. Helical representations of the frontier orbitals can be generated by linear combinations of the more localised orbitals from a symmetry-constrained calculation but they do not indicate π-conjugation. This work provides insights into the relationship between electronic structure and conformation in alkyne-linked conjugated oligomers.
Martin Peeks
Scientia Associate Professor
I am interested in understanding the fundamental chemistry of complex molecules and molecular systems.