Abstract
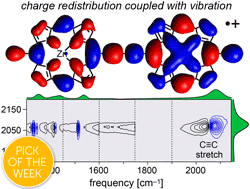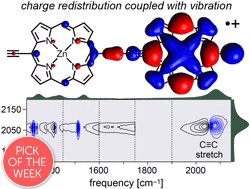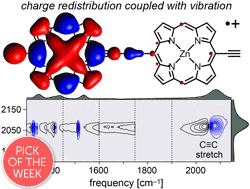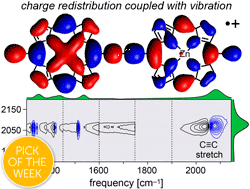
Break down of the Born–Oppenheimer approximation is caused by mixing of electronic and vibrational transitions in the radical cations of some conjugated polymers, resulting in unusually intense vibrational bands known as infrared active vibrations (IRAVs). Here, we investigate the mechanism of this amplification, and show that it provides insights into intramolecular charge migration. Spectroelectrochemical time-resolved infrared (TRIR) and two-dimensional infrared (2D-IR) spectroscopies were used to investigate the radical cations of two butadiyne-linked conjugated porphyrin oligomers, a linear dimer and a cyclic hexamer. The 2D-IR spectra reveal strong coupling between all the IRAVs and the electronic π–π* polaron band. Intramolecular vibrational energy redistribution (IVR) and vibrational relaxation occur within ∼0.1–7 ps. TRIR spectra show that the transient ground state bleach (GSB) and excited state absorption (ESA) signals have anisotropies of 0.31 ± 0.07 and 0.08 ± 0.04 for the linear dimer and cyclic hexamer cations, respectively. The small TRIR anisotropy for the cyclic hexamer radical cation indicates that the vibrationally excited polaron migrates round the nanoring on a time scale faster than the measurement, i.e. within 0.5 ps, at 298 K. Density functional theory (DFT) calculations qualitatively reproduce the emergence of the IRAVs. The first singlet (S1) excited states of the neutral porphyrin oligomers exhibit similar IRAVs to the radical cations, implying that the excitons have similar electronic structures to polarons. Our results show that IRAVs originate from the strong coupling of charge redistribution to nuclear motion, and from the similar energies of electronic and vibrational transitions.
Martin Peeks
Scientia Associate Professor
I am interested in understanding the fundamental chemistry of complex molecules and molecular systems.